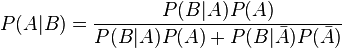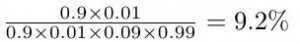Un article très intéressant est paru sur la BBC traitant des problèmes liés au diagnostic de maladies comme le cancer. Les problèmes naissent du fait que les les personnes saines sont plus nombreuses que les personnes malades (prévalence de la maladie basse) et que le test diagnostic peut être positif (i.e. la maladie est détectée) bien que le patient soit sain (taux de faux positifs différent de 0). Cela veut dire que même si le test est positif, les chances sont toujours hautes que la personne soit saine.
Exercice tiré de l’article
Quelle est la probabilité qu’une femme testée positive (i.e. malade) à une mammographie soit malade étant données les données suivantes ?
1. La probabilité qu’une femme ait le cancer du sein est de 1% (prévalence).
2. Si une femme a le cancer du sein, la probabilité que le test soit positif est de 90% (sensibilité du test).
3. Si une femme n’a pas le cancer du sein, la probabilité que le test soit quand même positif est de 9% (taux de fausse alerte).
Réponses possibles
a. 90%
b. 80%
c. 10%
d. 1%
Danse l’étude présentée par l’article, plus de trois quarts des médecins présentés avec cette question ont répondu faux ! Beaucoup (presque la moitié) ont répondu 90%, peut-être en raison du point 2 ci-dessus (probabilité que le test soit positif si la femme a le cancer). Cependant, la question est la probabilité que la femme ait le cancer si le test est positif ! Ce n’est pas la même chose !
Pour répondre à la question, il faudra rouvrir les cours de probabilité, en particulier il faut utiliser le théorème de Bayes.
Adaptons le théorème à notre cas particulier.
A : la femme a (vraiment) le cancer
B : le test est positif, le diagnostic suggère que la femme a le cancer
P(A | B) : étant donné que le test est positif (condition), la probabilité que la femme ait vraiment le cancer du sein
Les barres au-dessus des lettres indique la négation.
En remplaçant les probabilités données dans l’exercice dans la formule, nous obtenons ceci:
ce qui est environ égal à 10% ! bien loin des 90 % suggérés par les médecins.
Notez que le 99% (probabilité de ne pas avoir le cancer du sein) est le complémentaire du 1% (probabilité d’avoir le cancer du sein, sans a priori).
Si, comme moi, vous travaillez sur des jeux de données de grande dimensionalité et que vous en avez assez de parcourir la console à la recherche d’un summary lancé quelques dizaines commandes en arrière ou bien de relancer à chaque fois des commandes, vous allez aimer la commande capture.output(). Voici comment elle marche:
1. Tout d’abord, ouvrez le fichier de votre choix dans votre répertoire préféré. Par exemple, utilisez un chemin relatif à votre environnement de travail:
fileConn <- file(“regression/extracted_log.txt”)
2. Créez votre modèle:
model <- lm(variable_réponse ~ variable_explicative)
Le paramètre append de la fonction capture.output() est très utile si vous désirez ajouter la table en fin de fichier et ne pas écraser le contenu d’un fichier non vide. Ainsi, vous pouvez exporter une infinité de summary() dans le même fichier.
4. Fermez le fichier avec:
close(fileConn) #ici fileConn se refère au nom que vous avez donné à votre connexion plus haut.
Et voilà, ouvrez maintenant votre fichier texte pour regardez vos summary. Je trouve cette manière de faire plus propre et plus agréable.
1st Symposium of the Pôle Thématique de Recherche (PTR) Métabolisme-Nutrition-Vieillissement
LIMNA
Lausanne Integrative Metabolism and Nutrition Alliance
La journée commence avec gaieté avec une boutade d’un invité d’honneur, Grahame Hardie, de l’université de Dundee en Ecosse. A la question – rhétorique – d’en quoi consiste le métabolisme d’une cellule, le professeur réplique : à AMPK ! Ce qui semble extrêmement réductionniste devient en fait de plus en plus réaliste au fur et à mesure de la présentation. En effet, cette protéine kinase activée par l’AMP (AMP-activated kinase, AMPK) semble s’occuper de tout ce qui concerne la régulation de l’équilibre énergétique de la cellule.
Dans une cellule saine et non stressée, la concentration élevée d’ATP inhibe l’activité d’AMPK. En cas de stress, qui peut être causé par des poisons, une privation de glucose, la contraction musculaire, la prolifération rapide des cellules, la concentration d’ATP diminue et son hydrolyse provoque l’augmentation de la concentration d’ADP. L’équilibre entre l’ADP et l’AMP est tiré vers ce dernier, ce qui va finalement activer l’AMPK, simple non ? L’AMPK va ensuite activer le catabolisme pour refournir les stocks d’ATP.
Structure tridimensionnelle de l’AMPK : un résidu thréonine est phosphorylé par LKB1 et CaMKKβ (ce dernier activé par le Ca2+. L’AMPK contient des sous-unités α (1/2), β (1/2) et (1/2/3).
AMPK est le vrai chef d’orchestre du métabolisme cellulaire. Il va activer l’absorption du glucose, la glycolyse (ce processus qui transforme le glucose en pyruvate afin de produire de l’ATP), l’oxydation des acides gras, l’autophagie, la mitophagie, le métabolisme oxydatif et la biogenèse de mitochondries. L’AMPK a aussi des effets inhibiteurs, entre autres sur la synthèse du cholestérol, des triglycérides, des phospholipides, du glycogène, de l’ARN ribosomique, des protéines et la réplication de l’ADN. La levure du boulanger (Saccharomyces cerevisiae) a elle aussi un gène codant pour AMPK. Cet orthologue est nécessaire pour la croissance sur une source de carbone autre que le glucose ainsi que pour passer de la fermentation à un métabolisme oxydatif lorsque le glucose devient rare.
L’AMPK est régulé par des hormones et des cytokines. La leptin, cette hormone de la satiété, inhibe AMPK dans l’hypothalamus mais l’active dans les muscles.
L’AMP active AMPK de manière allostérique, c’est-à-dire que l’AMP va se lier sur AMPK à un endroit de la structure qui n’est pas le site active d’AMPK. Cette liaison favorise la phosphorylation activatrice de la thréonine d’AMPK par LKB1 et inhibe sa déphosphorylation, cette fois-ci par une phosphatase encore inconnue.
Le prof. Hardie parle ensuite de la berbérine, cette molécule dérivée d’une plante utilisée dans la médecine chinoise traditionnelle. C’est un inhibiteur mitochondrial et ce faisant, elle diminue la production d’ATP, augmente la concentration d’AMP, ce qui va in fine activer AMPK et aider l’homéostasie du glucose. Ce qui nous amène au sujet d’AMPK en tant que cible thérapeutique dans le diabète de type 2. En promouvant l’absorption du glucose et son oxydation, l’activation d’AMPK dans le muscle diminuerait la glycémie :
Médicaments qui inhibent les mitochondries et augmentent ainsi l’AMP cellulaire (metformine, phenformine, resveratrol, berbérine)
Médicaments se transformant en analogues de l’AMP :
AICAR → ZMP (analogue de l’AMP qui active AMPK
C13 → C2 (par des estérases cellulaires)
Médicaments se liant directement à AMPK : A-769662 (Abbott), 991 (Merck), MT 63-78 (Mercury), salicylate
La metformine inhibe le complexe I de la chaîne respiratoire des mitochondrie et ainsi, par l’augmentation de l’AMP cellulaire, inhibe AMPK.
AMPK : un suppresseur de tumeurs sous-régulé dans beaucoup de cancers
Non seulement AMPK joue un rôle central dans l’homéostasie du glucose, mais en plus il semble nous protéger du cancer. Ces deux effets à première vue complètement différents ne le sont en réalité pas vraiment lorsqu’on considère que les tumeurs sont très gourmandes en glucose. AMPK, par l’activation de l’absorption de glucose par les cellules va-t-il baisser la glycémie et ainsi priver les cellules cancéreuses de ce nutriment si chéri ? Plusieurs observations en tous cas soutiennent la candidature d’AMPK pour le poste de suppresseur de tumeurs :
Dans la maladie génétique rare du nom de syndrome de Peutz-Jeghers, LKB1, la kinase phosphorylant et activant AMPK, est mutée. Les patients développent des polypes intestinaux appelés hamartomes, ainsi que des tumeurs malignes en d’autres endroits du corps
L’activation d’AMPK inhibe la croissance cellulaire et cause l’arrêt du cycle cellulaire
Les patients diabétiques traités avec la metformine ont une incidence plus base de cancer
AMPK est sous-régulé dans les tumeurs, et ce par plusieurs mécanismes :
Perte génétique de LKB1
Réduction de l’expression de AMPK-α2
Hyperactivation du pathway Akt, e.g. perte de PTEN ou mutations activatrices dans la kinase PI-3
On comprend maintenant pourquoi le prof. Hardie a commencé sa présentation par dire que le métabolisme cellulaire se résumait à AMPK ! Même si ce n’est qu’une boutade, nous ne pouvons que restés émerveillés par cette protéine extraordinaire qui est au centre de l’homéostasie énergétique, non seulement de la cellule, mais de l’organisme entier.
Le séchage de gel en polyacrylamide permet la conservation sur le long terme de gels SDS-PAGE. Pour cela, il faut une solution pour le séchage des gels (4% glycérol, 20% éthanol), deux feuilles de cellophane, un bac, un gel, un couteau pour couper les puits du gel, une plaque en plastique, un cadre en plastique (cf. photos) et des clips.
Pendant toute la procédure, évitez de créer des bulles d’air entre les feuilles de cellophane et le gel !
1.Tremper le gel et les deux feuilles en cellophane dans le bac rempli de solution de séchage de gel (gel drying solution). Laisser 5 minutes au moins.
2. Poser la plaque sur la table.
3. Couvrir le cadre plein d’une feuille de cellophane imprégnée de solution de séchage de gel.
4. Poser le gel, dont les puits ont été coupés, sur la feuille de cellophane.
5. Recouvrir, en essayant d’éviter des bulles, le gel avec l’autre feuille de cellophane imprégnée de solution pour le séchage des gels.
6. Poser le cadre sur le tout et fermer avec des clips.
7. Laisser sécher près d’une fenêtre au moins 48h.
Solution de séchageCoupure des puitsCouverture du gel par une feuille de cellophane.
Couverture du gel par une feuille de cellophane. Une plaque se trouve sous mes mains et deux cadres se trouvent à l’arrière de l’image
Fermeture du sandwich avec des clipsSéchage à la fenêtre pendant au moins deux jours.
Souvent, lorsqu’on travaille avec une nouvelle construction plasmidique (construct), nous voulons vérifier que les bactéries qui ont incorporé le plasmide produisent bel et bien la protéine dont le gène se trouve sur le plasmide. Pour cela, il est conseillé d’effectuer un test d’expression de protéine à petite échelle avant de passer à une échelle plus grande, nécessaire à la production d’une quantité importante de protéine. Dans le cas où la protéine n’est pas exprimée, cela peut indiquer un problème avec le plasmide ou bien que la protéine est dégradée (en particulier dans le cas d’une expression dans des cellules eucaryotes, où le protéasome dégrade les protéines mal pliées) et il faut résoudre ce problème avant de passer à une expression à grande échelle.
Point de départ de l’expérience: colonies sur boîte d’agar.
Point d’arrivée: gels de polyacrylamide produits par SDS-PAGE et indiquant le degré d’expression de la protéine.
Marche à suivre
1. Choisir une ou plusieurs colonies en essayant d’éviter les colonies satellites (cf. lexique pour définition). Choisir plusieurs colonies augmente les chances d’obtenir au moins un résultat positif. Les étapes suivantes sont décrites pour une colonie.
2. Inoculer 4 tubes de culture de 12 ml contenant 2 ml de milieu de culture LB + antibiotique à partir d’une seule colonie. En outre, il est conseillé d’avoir un contrôle négatif et un contrôle positif pour l’expression. Pour le contrôle positif, utiliser un stock de glycérol dont on sait qu’il exprime une protéine (n’importe laquelle) et pour le contrôle négatif utiliser des bactéries non transformées. Enfin, utiliser un tube supplémentaire inoculé avec n’importe quelle colonie. Ce tube servira à mesur la densité optique (DO) à 600 nm.
3. Incuber avec rotation à 37°C pendant quelques heures jusqu’à ce que la densité optique à 600 nm du tube supplémentaire atteigne au moins 0.6. A ce moment-là, les 4 tubes inoculés avec la même colonie prennent des chemins différents:
a. Un tube, marqué iNI37, reste à 37°C sans aucun traitment. i va de 1 au nombre de colonies sélectionnées.
b. Un autre tube, marqué iI37, est induit à l’IPTG (1 mM de concentration finale) et reste à 37°C.
c. Un troisième tube, marqué iNI18, est déplacé à 18°C.
d. Le dernier tube, enfin, marqué iI18, est induit avec de l’IPTG et déplacé à 18°C. C’est ce dernier tube qui donne normalement l’expression maximale, en raison de l’induction et du déplacement à 18°C. A cette dernière température, les bactéries cessent de se multiplier et peuvent focaliser leur énergie sur la production de la protéine d’intérêt.
4. Laisser incuber au moins 3,5 heures aux températures respectives. Si c’est très tard et que vous voulez rentrer à la maison absolument, vous pouvez aussi laisser incuber du jour au lendemain.
5. Après l’incubation, transférer 1 ml de culture dans un tube eppendorf de 1,5 ml.
6. Centrifuger 10 min à 15’000 rpm.
7. Jeter le surnageant.
8. Resolubiliser le culot dans 200 µl de BugBuster. Cette solution va lyser les cellules.
9. Centrifuger 10 min à 15’000 rpm.
10. Transférer le surnageant (la partie soluble) dans un autre eppendorf de 1,5 ml.
11. Aliquoter 30 µl de partie soluble dans un autre tube eppendorf contenant 30 µl de solution Lämmli 2x (solution de chargement pour le SDS-PAGE).
12. Resuspendre le culot (la partie insoluble) dans 50 µl de solution Lämmli 2x.
13. Chauffer les tubes contenant le Lämmli à 95°C pendant 5 min et charger dans des gels de polyacrylamide.
Il est aussi possible d’utiliser des erlenmeyer au lieu des tubes de culture.
Lexique
Colonie satellite: colonie de bactéries non transformées qui réussit à pousser sur de l’agar contenant de l’antibiotique en raison de la dégradation de l’antibiotique par une colonie voisine qui exprime la résistance à l’antibiotique et dégrade l’antibiotique qui l’entoure.
Cette solution est utilisée après avoir coloré un gel de polyacrylamide au bleu de Coomassie. Il faut faire baigner le gel dans cette solution, chauffer 1 min au micro-ondes et laisser décolorer sur une plateforme en mouvement pendant quelques dizaines de minutes. Ensuite, jeter le liquide coloré dans l’évier et répéter jusqu’à obtention d’un gel suffisamment décoloré (jusqu’à ce que seules les bandes de protéines restent bleues).
Pour 2 litres:
– 1 litre d’EtOH (éthanol pur ou éthanol avec 5% de méthanol)
– 800 ml d’H20
– 200 ml d’acide éthanoïque (= acide acétique)
Si l’acide éthanoïque est à 80% -> verse 850 ml d’H20 et 250 ml d’acide éthanoïque à la place.
27.02.14 Les plasmides ont été ajoutés aux bactéries et se lient à la surface de la membrane plasmique.
27.02.14 Les plasmides ont été ajoutés aux bactéries et se lient à la surface de la membrane plasmique.27.02.14 Une bouteille d’agar solide, avant d’être fondue.27.02.14 Des boîtes de Pétri d’agar en train de sécher autour d’une flamme de bec Bunsen.27.02.14 Les bactéries, après transformation, sont incubées avec rotation à 37°C pour leur laisser le temps d’exprimer la résistance à l’antibiotique.50 ul de bactéries sont étalées sur les boîtes de Pétri autour du feu.Les boîtes de Pétri sont incubées du jour au lendemain à 37°C dans le thermostat.
Lausanne, le 27 février 2014
La transformation des bactéries avec un plasmide est utile si nous voulons exprimer un gène dans une souche de bactéries, par exemple un gène codant pour une protéine. Dans mon travail au Laboratoire de biologie structurale et biophysique à l’EPFL, je suis souvent amené à transformer des bactéries avec un plasmide. Aujourd’hui par exemple, j’aimerais transformer 4 plasmides contenant chacun le gène de la protéine X avec une mutation différente. J’aimerais transformer ces plasmides dans la souche E. coli B834(De3) parce que cette dernière se prête particulièrement bien à l’expression de protéines. J’utilise les bactéries en quelque sorte comme des petites usines pour produire ma protéine d’intérêt. D’autres souches de E. coli se prêtent mieux à d’autres fins, e.g. la souche DH5α se prête bien à la production de plasmide afin de l’extraire par préparation de plasmide (miniprep, midiprep ou maxiprep) pour augmenter notre stock de plasmide.
Aujourd’hui, je procèderai à une transformation chimique des bactéries. Il existe aussi la transformation électrique (par électroporation) des bactéries, discutée dans un autre post.
Marche à suivre
1. Sortir les bactéries compétentes B834(De3) du congélateur à – 80 °C et les faire dégeler sur de la glace. Faire de même avec les plasmides stockés à – 20 °C.
2. Pour chaque transformation, transférer 50 µl de bactéries compétentes dans un tube eppendorf de 1,5 ml.
3. Ajouter 2 µl de plasmide à une concentration de 15 ng/µl. La quantité finale de plasmide doit se situer entre 10 ng et 100 ng. Dans le cas présent, la mass finale de plasmide est de 30 ng.
4. Incuber le mélange de plasmide et de bactéries compétentes pendant 30 minutes sur la glace.
5. Pendant les 30 minutes d’incubation, nous allons préparer des boîtes d’agar contenant de l’antibiotique (ici de l’ampicilline). Si les boîtes sont déjà prêtes, les sortir du frigo et les mettre dans le thermostat à 37°C. Dans le cas contraire, nous allons préparer des boîtes d’agar fraîches. D’abord, faire fondre l’agar au micro-ondes. Perso, je fais fondre graduellement, i.e. pendant 2 minutes séparées d’un coup d’oeil pour monitorer l’avancement de la fusion et l’état du micro-ondes. Attention à que le bouchon ne soit pas fortement fermé (tight). Le bouchon doit pouvoir danser sur la bouteille, i.e. être lâche (loose), afin que l’air puisse sortir de la bouteille pendant l’augmentation de température (rappel de la loi des gaz parfaits: PV = nRT -> si la température augmente et que la bouteille est fermée (V constant), la pression dans la bouteille augmente et celle-ci risque d’exploser!). Après fusion de l’agar, en verser une quantité suffisante dans des tubes falcon de 50 ml (environ 20 ml par boîte de Pétri). Attendre que la température baisse suffisamment avant d’ajouter l’antibiotique. En effet, si l’antibiotique est ajouté trop tôt, la température élevée risque de le détruire. D’un autre côté, si nous attendons trop avant d’ajouter l’antibiotique, l’agar risque de se solidifier. Les étapes suivantes se passent près d’une flamme au bec Bunsen afin de ne pas contaminer l’agar avec des bactéries de l’air. Aujourd’hui, je prépare 5 boîtes d’agar avec antibiotique et je verse donc 100 ml d’agar dans 2 tubes falcons de 50 ml. Après que la température soit descendue suffisamment (un bon indice est qu’en touchant le tube avec l’avant bras, la sensation ne doit pas être douloureuse), j’ajoute 50 µl d’antibiotique 1000 x dans chaque tube. Je verse ensuite l’agar liquide dans les boîtes autour de la flamme et je les laisse ouvertes afin qu’elles sèchent.
6. Après les 30 minutes d’incubation sur glace, nous passons au choc thermique: chauffer les bactéries pendant 5 minutes à 42 °C sur un bloc chauffant.
7. Remettre les tubes sur glace pendant 2 minutes.
8. Ajouter 1 ml de milieu LB dans le tube eppendorf de 1,5 ml et transférer le tout (1,057 ml) dans des tubes de culture de 12 ml à incuber pendant 30-60 min à 37 °C à 220 rpm. Cette période de temps permet aux bactéries d’exprimer la résistance à l’antibiotique codée sur le plasmide.
9. Etaler 50 µl sur les boîtes d’agar contenant l’antibiotique. Seules les bactéries ayant incorporé l’antibiotique formeront des colonies pendant la nuit.
10. Incuber du jour au lendemain à 37°C dans le thermostat.
Lausanne, le 28 février 2014
9. Vérifier la présence de colonies dans les boîtes sur lesquelles des bactéries transformées ont été étalées. Vérifier que le contrôle négatif, la boîte sur laquelle des bactéries non transformées ont été étalées, ne contienne pas de colonies.
28.02.14 Des colonies ont poussé pendant la nuit sur toutes les boîtes sur lesquelles des bactéries transformées ont été étalées. Le contrôle négatif, sur laquelle des bactéries non transformées ont été étalées, ne contient pas de colonies.
ingrédients
– des bactéries “compétentes”, i.e. facilement transformables par de l’ADN exogène. Les bactéries doivent soit être sensibles à un antibiotique, soit être auxotrophes.
– de l’agar en poudre
– un plasmide portant un gène de sélection. e.g. un gène produisant de la beta-lactamase donnant la résistance à l’antibiotique ampicilline ou un gène dont le produit permet la synthèse d’un nutriment rendant les bactéries qui l’expriment prototrophes.
– en fonction du gène de sélection: antibiotique ou milieu sélectif pour bactéries prototrophes.
L’idée de créer un site internet dédiés à mes activités scientifiques est née de mes expériences nombreuses en recherche scientifique et dans l’enseignement. C’est un moyen de résumer la richesse que m’ont apportée ces expériences d’ici et d’ailleurs. Qui plus est, créer un site internet était en soi un défi que je voulais relever par curiosité et un prétexte pour apprendre de nouvelles habiletés et de nouveaux langages, tels que HTML, PHP, etc.
Dans ce site, encore à ses balbutiements, j’ai l’intention de poster des vidéos d’expérience en laboratoire afin de partager des techniques avec d’autres scientifiques, des vidéos éducatives dans les branches que j’ai enseignées à l’université et d’autres qui me tiennent à coeur: statistique, programmation, botanique, physique et autres. Une partie du site sera aussi dédiée à mes activités de bénévolat dans le cadre de projet environnementaux pour l’entretien des forêts.